\textrm{Sr}_{2}\textrm{ScSbO}_{6}, RT
Contents
Target
Perform previous calculations, as needed
Obtain the control file,
.pcr, and understand it
Steps
Take the structure from the paper
Take the Sb to the origin
Convert non-standard to standard
AMPLIMODES needs the Transformation Matrix (TM)
AMPLIMODES with non-standard: Transformation Matrix
AMPLIMODES with standard: Transformation Matrix
AMPLIMODES for FullProf with non-standard/standard
Comparison of the control file (
.pcr/.new) between co-ordinates and modesComments
Take the structure from the paper¶
# P 1 21/n 1
014
5.6915 5.6778 8.0244 90.000000 90.03 90.000000
6
Sb 1 - 0.000000 0.500000 0.000000
Sc 1 - 0.500000 0.000000 0.000000
Sr 1 - 0.002100 0.009900 0.248700
O 1 - 0.261500 0.268000 0.028000
O 2 - 0.274000 0.262000 0.477000
O 3 - 0.949300 0.494400 0.245600
Take the Sb to the origin¶
Copy structure
Use EQUIVSTRU
Result
# P 1 21/n 1
014
5.6915 5.6778 8.0244 90.00 90.03 90.00
6
Sb 1 - 0.000000 0.000000 0.000000
Sc 1 - 0.500000 -0.500000 0.000000
Sr 1 - 0.002100 -0.490100 0.248700
O 1 - 0.261500 -0.232000 0.028000
O 2 - 0.274000 -0.238000 0.477000
O 3 - 0.949300 -0.005600 0.245600
Convert non-standard to standard¶
Copy structure
Use SETSTRU
Result
# P 1 21/c 1
014
8.0244 5.6778 9.8355 90.00 144.64 90.00
6
Sb 1 2a 0.000000 0.000000 0.000000
Sc 1 2b -0.500000 -0.500000 -0.500000
Sr 1 4e 0.246600 -0.490100 -0.002100
O 1 4e -0.233500 -0.232000 -0.261500
O 2 4e 0.203000 -0.238000 -0.274000
O 3 4e -0.703700 -0.005600 -0.949300
AMPLIMODES needs the Transformation Matrix {TM}¶
Parent Phase
# F m -3 m
225
8.087481 8.087481 8.087481 90.00000 90.00000 90.00000
4
Sb 1 4a 0.000000 0.000000 0.000000
Sc 1 4b 0.500000 0.500000 0.500000
Sr 1 8c 0.250000 0.250000 0.250000
O 11 24e 0.254100 0.000000 0.000000
At least, 3 ways to obtain the Transformation Matrix:
Calculate by hand: Previous Information
Use PSEUDO: be careful, only admits standard settings You can obtain it also for the non-standard setting
Calculate by STRUCTURE RELATIONS It is also reachable from AMPLIMODES
1. Calculate by hand: Previous Information
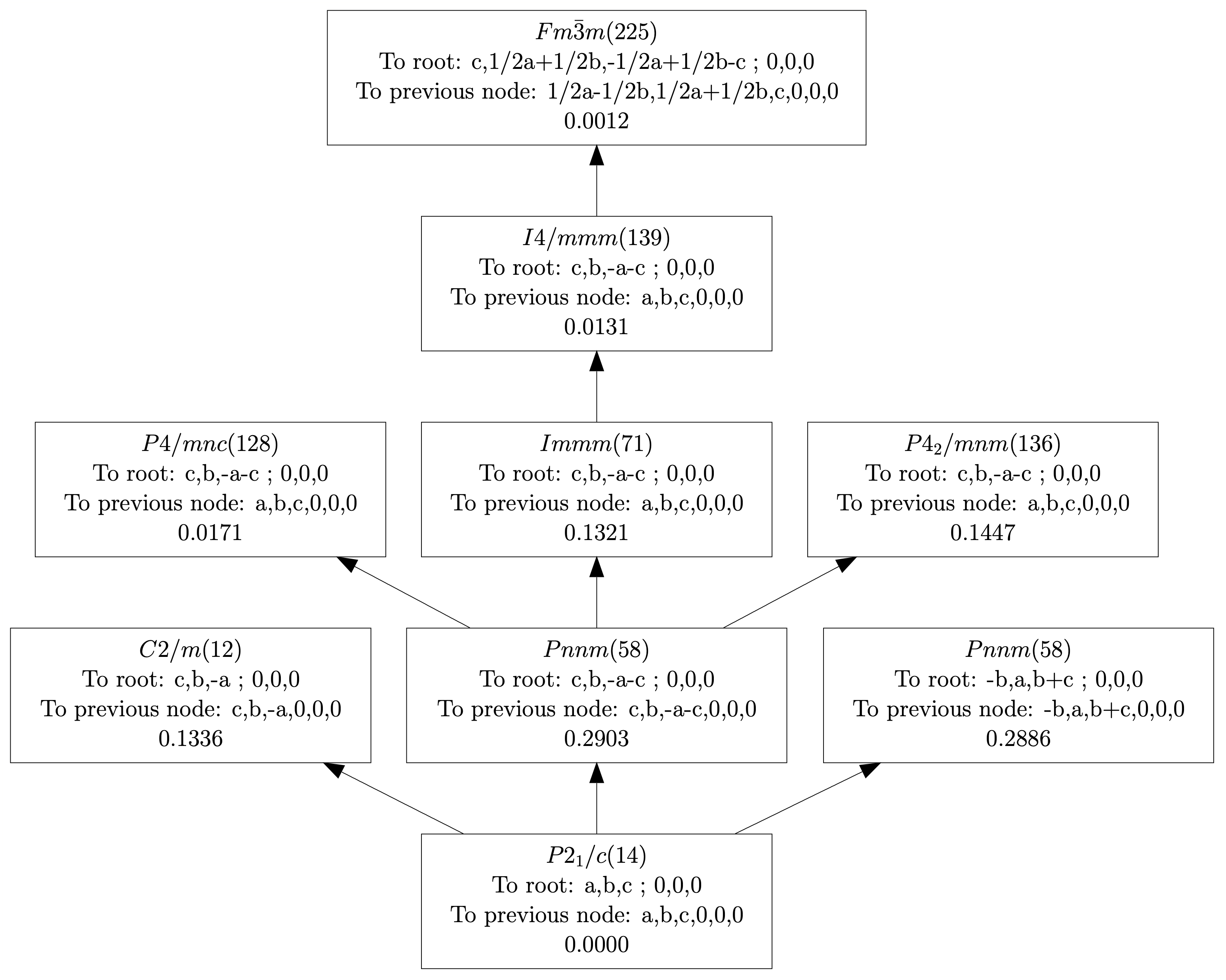
3. Calculate by STRUCTURE RELATIONS
Transformation Matrix (P,p): (b,1/2a-1/2c,-1/2a-b-1/2c;0,0,0)
Transformation matrix:
[ 0 1/2 -1/2 ] [ 0]
[ 1 0 -1 ] [ 0]
[ 0 -1/2 -1/2 ] [ 0]
You can check that both matrices give the same final result…
Result, for the non-standard
Transformation Matrix (P,p): (1/2a-1/2b,1/2a+1/2b,c;0,0,0)
Result, for the standard
Transformation Matrix (P,p): (b,1/2a-1/2c,-1/2a-b-1/2c;0,0,0)
Transformation Matrix (P,p): (c,1/2a+1/2b,-1/2a+1/2b-c;0,0,0)
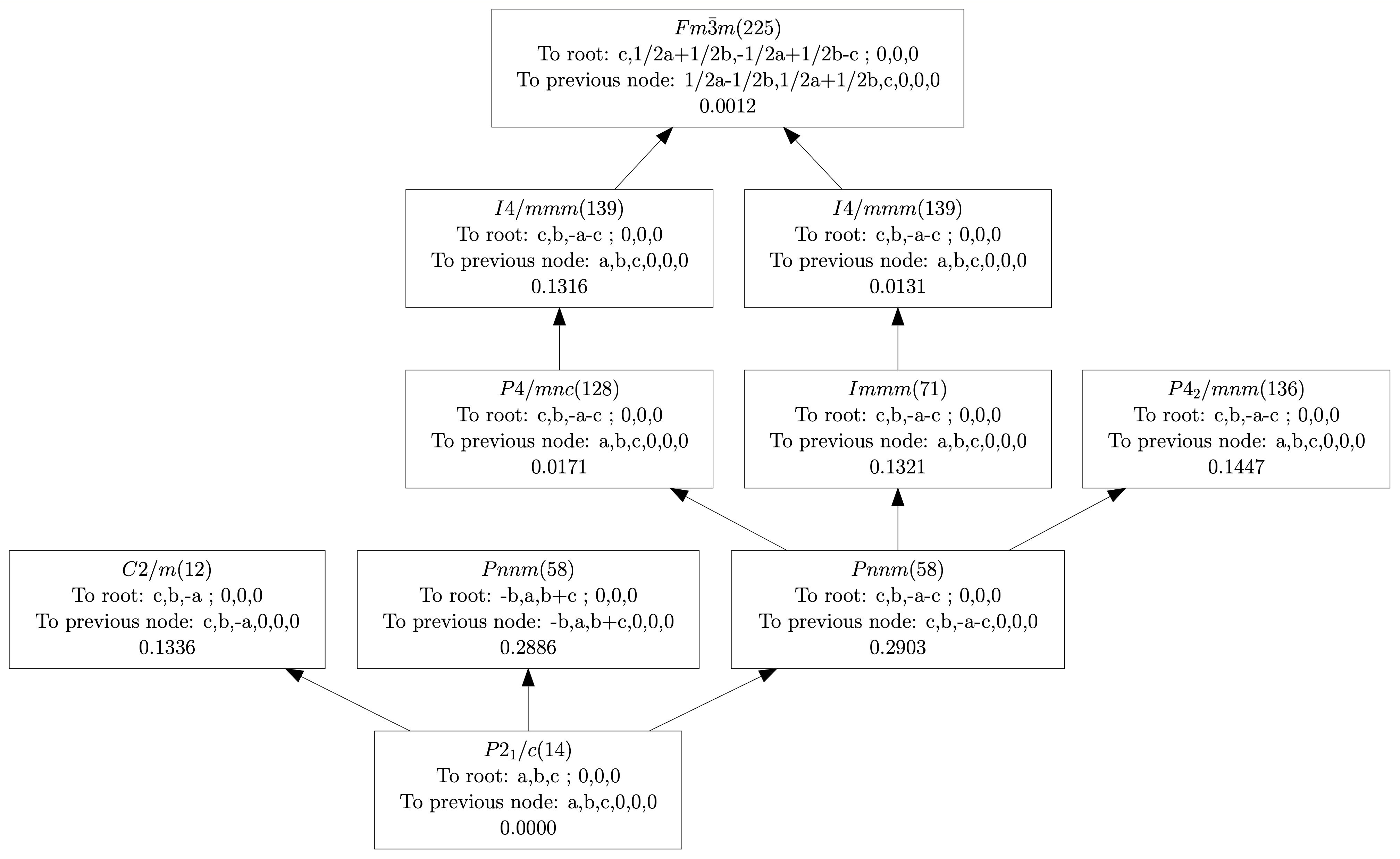
AMPLIMODES with non-standard: Transformation Matrix¶
# F m -3 m
225
8.087481 8.087481 8.087481 90.00000 90.00000 90.00000
4
Sb 1 4a 0.000000 0.000000 0.000000
Sc 1 4b 0.500000 0.500000 0.500000
Sr 1 8c 0.250000 0.250000 0.250000
O 11 24e 0.254100 0.000000 0.000000
# P 1 21/n 1
014
8.0244 5.6778 9.8355 90.00 144.64 90.00
6
Sb 1 2a 0.000000 0.000000 0.000000
Sc 1 2b -0.500000 -0.500000 -0.500000
Sr 1 4e 0.246600 -0.490100 -0.002100
O 1 4e -0.233500 -0.232000 -0.261500
O 2 4e 0.203000 -0.238000 -0.274000
O 3 4e -0.703700 -0.005600 -0.949300
Transformation matrix:
[ 0 1/2 -1/2 ] [ 0]
[ 1 0 -1 ] [ 0]
[ 0 -1/2 -1/2 ] [ 0]
b,1/2a-1/2c,-1/2a-b-1/2c; 0 0 0
AMPLIMODES with standard: Transformation Matrix¶
# F m -3 m
225
8.087481 8.087481 8.087481 90.00000 90.00000 90.00000
4
Sb 1 4a 0.000000 0.000000 0.000000
Sc 1 4b 0.500000 0.500000 0.500000
Sr 1 8c 0.250000 0.250000 0.250000
O 11 24e 0.254100 0.000000 0.000000
# P 1 21/n 1
014
5.6915 5.6778 8.0244 90.00 90.03 90.00
6
Sb 1 - 0.000000 0.000000 0.000000
Sc 1 - 0.500000 -0.500000 0.000000
Sr 1 - 0.002100 -0.490100 0.248700
O 1 - 0.261500 -0.232000 0.028000
O 2 - 0.274000 -0.238000 0.477000
O 3 - 0.949300 -0.005600 0.245600
Transformation matrix:
[ 1/2 1/2 0 ] [ 0]
[ -1/2 1/2 0 ] [ 0]
[ 0 0 1 ] [ 0]
1/2a+1/2b,1/2a-1/2b,c; 0 0 0
AMPLIMODES for FullProf with non-standard/standard¶
# F m -3 m
225
8.087481 8.087481 8.087481 90.00000 90.00000 90.00000
4
Sb 1 4a 0.000000 0.000000 0.000000
Sc 1 4b 0.500000 0.500000 0.500000
Sr 1 8c 0.250000 0.250000 0.250000
O 11 24e 0.254100 0.000000 0.000000
# P 1 21/n 1
014
5.6915 5.6778 8.0244 90.00 90.03 90.00
6
Sb 1 - 0.000000 0.000000 0.000000
Sc 1 - 0.500000 -0.500000 0.000000
Sr 1 - 0.002100 -0.490100 0.248700
O 1 - 0.261500 -0.232000 0.028000
O 2 - 0.274000 -0.238000 0.477000
O 3 - 0.949300 -0.005600 0.245600
Transformation matrix:
[ 1/2 1/2 0 ] [ 0]
[ -1/2 1/2 0 ] [ 0]
[ 0 0 1 ] [ 0]
1/2a-1/2b,1/2a+1/2b,c; 0 0 0
Comparison of the control file (.pcr/.new) between co-ordinates and modes¶
!Nat Dis Ang Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More
6 0 0 1.0 0.0 0.0 0 0 0 0 0 875.885 0 7 1
!
!Jvi Jdi Hel Sol Mom Ter Brind RMua RMub RMuc Jtyp Nsp_Ref Ph_Shift N_Domains
0 3 0 0 0 0 1.0000 0.0000 0.0000 0.0000 1 0 0 0
!
! Max_dst(dist) (angles) Bond-Valence Calc.
3.2000 3.2000 BVS
! N_cations N_anions Tolerance(%) / Name or cations/ and Anions
3 1 0.00
SR+2 SC+3 SB+5
O-2
!
!
P 1 21/N 1 <--Space group symbol
!Atom Typ X Y Z Biso Occ In Fin N_t Spc /Codes
Sb SB 0.00000 0.50000 0.00000 0.14020 0.50000 0 0 0 1
0.00 0.00 0.00 271.00 0.00
Sc SC 0.50000 0.00000 0.00000 0.14020 0.50000 0 0 0 2
0.00 0.00 0.00 271.00 0.00
Sr SR 0.00023 0.00977 0.25025 0.32583 1.00000 0 0 0 3
131.00 141.00 151.00 261.00 0.00
O1 O 0.25933 0.27162 0.02536 0.49182 1.00000 0 0 0 4
161.00 171.00 181.00 251.00 0.00
O2 O 0.27538 0.26338 0.47468 0.49182 1.00000 0 0 0 4
191.00 201.00 211.00 251.00 0.00
O3 O 0.94813 0.49532 0.24404 0.49182 1.00000 0 0 0 4
221.00 231.00 241.00 251.00 0.00
!Nat Dis Ang Pr1 Pr2 Pr3 Jbt Irf Isy Str Furth ATZ Nvk Npr More
6 0 0 1.0 0.0 0.0 6 0 0 0 12 875.885 0 7 1
!
!Jvi Jdi Hel Sol Mom Ter Brind RMua RMub RMuc Jtyp Nsp_Ref Ph_Shift N_Domains
0 3 0 0 0 0 1.0000 0.0000 0.0000 0.0000 1 0 0 0
!
! Max_dst(dist) (angles) Bond-Valence Calc.
3.2000 3.2000 BVS
! N_cations N_anions Tolerance(%) / Name or cations/ and Anions
3 1 0.00
SR+2 SC+3 SB+5
O-2
!
!
P 1 21/n 1 <--Space group symbol
!Atom Typ X Y Z Biso Occ In Fin N_t Spc /Codes
Sb1 SB 0.000000 0.000000 0.000000 0.500000 0.500000 0 0 0 1
0.00 0.00 0.00 0.00 0.00
Sc1 SC 0.500000 0.000000 0.000000 0.500000 0.500000 0 0 0 1
0.00 0.00 0.00 0.00 0.00
Sr1 SR 0.750000 0.000000 0.500000 0.500000 1.000000 0 0 0 1
0.00 0.00 0.00 0.00 0.00
O11 O 0.745900 0.254100 0.745900 0.500000 1.000000 0 0 0 1
0.00 0.00 0.00 0.00 0.00
O11_2 O 0.254100 0.500000 0.500000 0.500000 1.000000 0 0 0 1
0.00 0.00 0.00 0.00 0.00
O11_3 O 0.745900 0.754100 0.245900 0.500000 1.000000 0 0 0 1
0.00 0.00 0.00 0.00 0.00
! Polarisation Vectors of Symmetry Modes for each atom
V_MODES 30
! Nm Atm Irrep Vx Vy Vz Coeff
1 O11 GM1+ -0.035694 0.035694 -0.035694 1.00
1 O11_2 GM1+ 0.035694 0.000000 0.000000 1.00
1 O11_3 GM1+ -0.035694 0.035694 -0.035694 1.00
2 O11 GM3+ -0.025240 0.025240 -0.025240 1.00
2 O11_2 GM3+ -0.050479 0.000000 0.000000 1.00
2 O11_3 GM3+ -0.025240 0.025240 -0.025240 1.00
3 O11 GM4+ -0.030912 0.000000 0.000000 1.00
3 O11_2 GM4+ -0.061824 0.000000 -0.061824 1.00
3 O11_3 GM4+ -0.030912 0.000000 0.000000 1.00
4 Sr1 GM5+ 0.061824 0.000000 0.000000 1.00
5 Sr1 GM5+ -0.087432 0.000000 -0.087432 1.00
6 O11 GM5+ 0.030912 0.000000 0.000000 1.00
6 O11_2 GM5+ -0.061824 0.000000 -0.061824 1.00
6 O11_3 GM5+ 0.030912 0.000000 0.000000 1.00
7 O11 GM5+ -0.043716 -0.043716 -0.043716 1.00
7 O11_2 GM5+ 0.000000 0.000000 0.000000 1.00
7 O11_3 GM5+ -0.043716 -0.043716 -0.043716 1.00
8 O11 X2+ -0.043716 0.043716 -0.043716 1.00
8 O11_2 X2+ 0.000000 0.000000 0.000000 1.00
8 O11_3 X2+ 0.043716 -0.043716 0.043716 1.00
9 O11 X3+ 0.043716 0.043716 0.043716 1.00
9 O11_2 X3+ 0.000000 0.000000 0.000000 1.00
9 O11_3 X3+ -0.043716 -0.043716 -0.043716 1.00
10 Sr1 X5+ 0.000000 -0.087432 0.000000 1.00
11 O11 X5+ -0.043716 0.000000 0.000000 1.00
11 O11_2 X5+ 0.000000 0.000000 0.000000 1.00
11 O11_3 X5+ 0.043716 0.000000 0.000000 1.00
12 O11 X5+ 0.000000 0.000000 0.000000 1.00
12 O11_2 X5+ 0.000000 0.087432 0.000000 1.00
12 O11_3 X5+ 0.000000 0.000000 0.000000 1.00
!Amplitudes of Symmetry Modes
A_MODES 12 2
Max_Amplitude 2.0
A1_GM1+ -0.242338 1.00
A2_GM3+ -0.002972 1.00
A3_GM4+ -0.822497 1.00
A4_GM5+ 0.021028 1.00
A5_GM5+ -0.024019 1.00
A6_GM5+ 0.002426 1.00
A7_GM5+ -0.037172 1.00
A8_X2+ -0.031453 1.00
A9_X3+ 0.374577 1.00
A10_X5+ -0.113230 1.00
A11_X5+ 0.057188 1.00
A12_X5+ 0.064050 1.00
Comments¶
The structure is fixed: you refine the amplitude values The structure is the parent phase Result form AMPLIMODES:
Transformed high symmetry structure in the subgroup basis Reference Structure
Symmetry mode analysis High symmetry structure 225 8.087481 8.087481 8.087481 90.00000 90.00000 90.00000 4 Sb 1 4a 0.000000 0.000000 0.000000 Sc 1 4b 0.500000 0.500000 0.500000 Sr 1 8c 0.250000 0.250000 0.250000 O 11 24e 0.254100 0.000000 0.000000 Transformation matrix [ 0 1/2 -1/2 ] [ 0] [ 1 0 -1 ] [ 0] [ 0 -1/2 -1/2 ] [ 0] Transformed high symmetry structure in the subgroup basis Reference Structure 014 8.087481 5.718712 9.905101 90.000000 144.735611 90.000000 6 Sb 1 2a 0.000000 0.000000 0.000000 Sc 1 2b 0.500000 0.000000 0.000000 Sr 1 4e 0.750000 0.000000 0.500000 O 11 4e 0.745900 0.254100 0.745900 O 11_2 4e 0.254100 0.500000 0.500000 O 11_3 4e 0.745900 0.754100 0.245900
From AMPLIMODES for FullProf:
P 1 21/n 1 <--Space group symbol !Atom Typ X Y Z Biso Occ In Fin N_t Spc /Codes Sb1 SB 0.000000 0.000000 0.000000 0.500000 0.500000 0 0 0 1 0.00 0.00 0.00 0.00 0.00 Sc1 SC 0.500000 0.000000 0.000000 0.500000 0.500000 0 0 0 1 0.00 0.00 0.00 0.00 0.00 Sr1 SR 0.750000 0.000000 0.500000 0.500000 1.000000 0 0 0 1 0.00 0.00 0.00 0.00 0.00 O11 O 0.745900 0.254100 0.745900 0.500000 1.000000 0 0 0 1 0.00 0.00 0.00 0.00 0.00 O11_2 O 0.254100 0.500000 0.500000 0.500000 1.000000 0 0 0 1 0.00 0.00 0.00 0.00 0.00 O11_3 O 0.745900 0.754100 0.245900 0.500000 1.000000 0 0 0 1 0.00 0.00 0.00 0.00 0.00Differences:
Jbt=6Furth=12(depending on the case!)vector polarization list: you could generate it by hand you have to go to the Detailed Information, and further…
Results from AMPLIMODES
Symmetry mode analysis
High symmetry structure
Setting used: FM-3MTransformation Matrix: a,b,c
225 8.087481 8.087481 8.087481 90.00000 90.00000 90.00000 4 Sb 1 4a 0.000000 0.000000 0.000000 Sc 1 4b 0.500000 0.500000 0.500000 Sr 1 8c 0.250000 0.250000 0.250000 O 1 24e 0.254100 0.000000 0.000000
Low symmetry structure
Setting used: P 1 21/n 1Transformation Matrix: c,b,-a-c
014 5.6915 5.6778 8.0244 90.00 90.03 90.00 6 Sb 1 - 0.000000 0.000000 0.000000 Sc 1 - 0.500000 -0.500000 0.000000 Sr 1 - 0.002100 -0.490100 0.248700 O 1 - 0.261500 -0.232000 0.028000 O 2 - 0.274000 -0.238000 0.477000 O 3 - 0.949300 -0.005600 0.245600
Transformation matrix
[ 1/2 1/2 0 ] [ 0] [ -1/2 1/2 0 ] [ 0] [ 0 0 1 ] [ 0]
Transformed high symmetry structure in the subgroup basis
Reference Structure014 8.087500 5.718726 9.905124 90.000000 144.735626 90.000000 6 Sb 1 2a 0.000000 0.000000 0.000000 Sc 1 2b 0.500000 0.000000 0.000000 Sr 1 4e 0.250000 0.500000 0.000000 O 1 4e 0.745900 0.254100 0.745900 O 1_2 4e 0.254100 0.500000 0.500000 O 1_3 4e 0.254100 0.254100 0.254100
Atom pairings and distances
| Atom Mappings | |||||
|---|---|---|---|---|---|
| WP | Atom | Reference Struc. | Atom | Low Sym Struc. | |
| 2a | (0,0,0) | Sb1 | (0.000000,0.000000,0.000000) | Sb1 | (0.000000,0.000000,0.000000) |
| 2b | (1/2,0,0) | Sc1 | (0.000000,0.000000,0.500000) | Sc1 | (0.000000,0.000000,0.500000) |
| 4e | (x,y,z) | Sr1 | (0.000000,0.500000,0.250000) | Sr1 | (-0.997900,0.509900,-0.751300) |
| 4e | (x,y,z) | O1 | (-0.745900,0.254100,0.000000) | O2 | (-0.774000,0.262000,0.023000) |
| 4e | (x,y,z) | O1_2 | (-0.500000,0.500000,-0.245900) | O3 | (-0.550700,0.505600,-0.254400) |
| 4e | (x,y,z) | O1_3 | (-0.254100,0.254100,0.000000) | O1 | (-0.261500,0.232000,-0.028000) |
| WP | Atom | Atomic Displacements | ||||
|---|---|---|---|---|---|---|
| ux | uy | uz | |u| | |||
| 2a | (0,0,0) | Sb1 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| 2b | (1/2,0,0) | Sc1 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| 4e | (x,y,z) | Sr1 | 0.0021 | 0.0099 | -0.0013 | 0.0588 |
| 4e | (x,y,z) | O1 | -0.0281 | 0.0079 | 0.0230 | 0.2499 |
| 4e | (x,y,z) | O1_2 | -0.0507 | 0.0056 | -0.0085 | 0.2997 |
| 4e | (x,y,z) | O1_3 | -0.0074 | -0.0221 | -0.0280 | 0.2628 |
NOTE: ux, uy and uz are given in relative units. |u| is the absolute distance given in Å
Maximum atomic displacement in the distortion, Δ: 0.2997 Å
Total distortion amplitude: 0.9482 Å
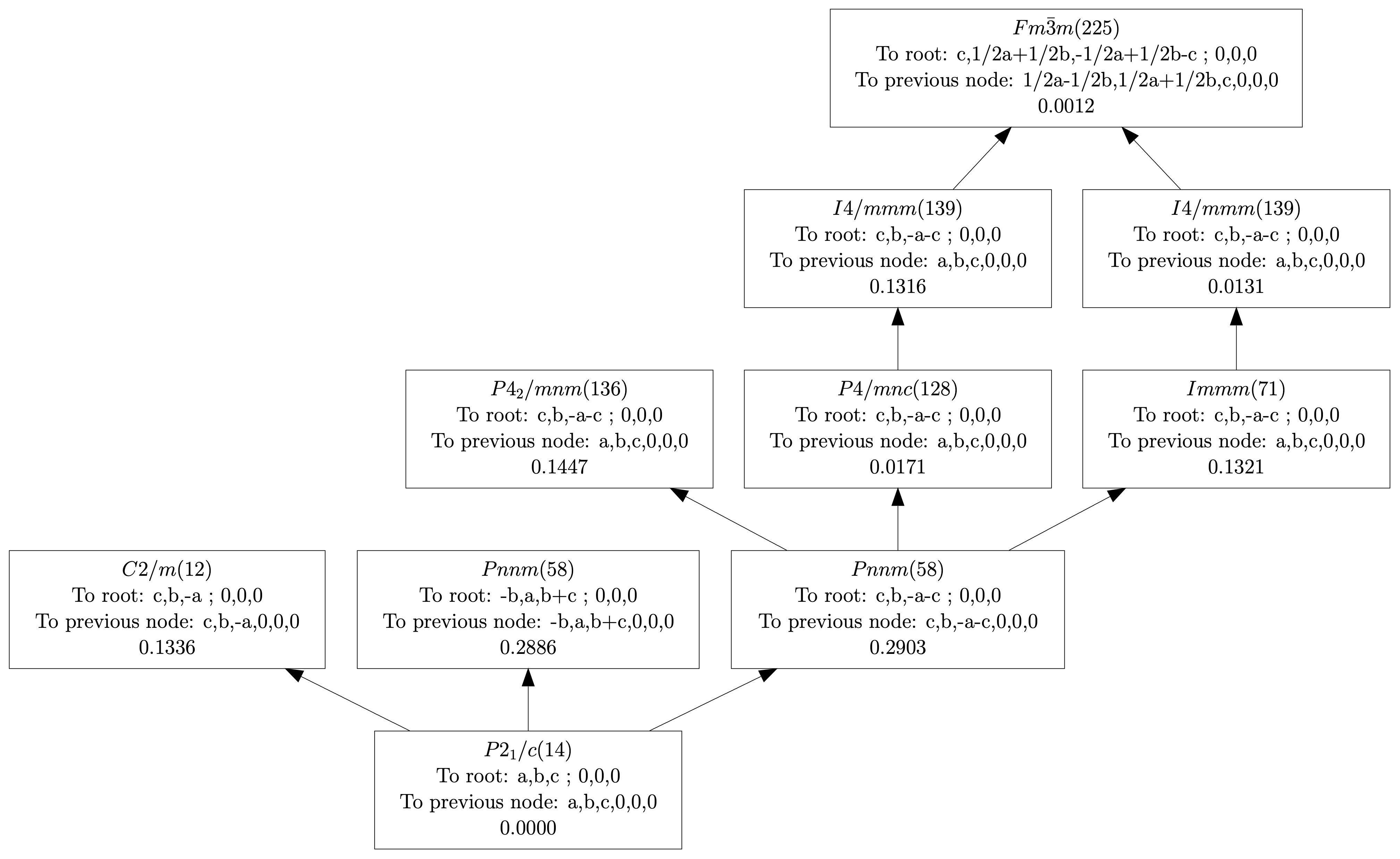
Symmetry Modes Summary
| Atoms | WP | Modes |
| O1 | 24e | GM1+(1) GM3+(1) GM4+(1) GM5+(2) X2+(1) X3+(1) X5+(2) |
| Sr1 | 8c | GM5+(2) X5+(1) |
Summary of Amplitudes
Warning: Amplitudes are given for modes normalized within the primitive unit cell of the distorted structure. Under this normalization, mode amplitudes in distorted structures with different multiplication of their primitive unit cell are not directly comparable.
| K-vector | Irrep | Direction | Isotropy Subgroup | Dimension | Amplitude (Å) |
| (0,0,0) | GM1+ | (a) | Fm-3m (225) | 1 | 0.2423 |
| (0,0,0) | GM3+ | (a,0) | I4/mmm (139) | 1 | 0.0030 |
| (0,0,0) | GM4+ | (a,a,0) | C2/m (12) | 1 | 0.8225 |
| (0,0,0) | GM5+ | (-b,a,-a) | C2/m (12) | 4 | 0.0491 |
| (0,1,0) | X2+ | (0,a,0) | P4_2/mnm (136) | 1 | 0.0315 |
| (0,1,0) | X3+ | (0,a,0) | P4/mnc (128) | 1 | 0.3746 |
| (0,1,0) | X5+ | (a,a,0,0,a,-a) | Pnnm (58) | 3 | 0.1421 |
Global distortion: 0.9482 Å
Normalized basis symmetry modes
The modes are normalized to the reference structure unit cell and are given as relative displacements in this cell.K-vector: GM = (0,0,0)
Irrep GM1+
GM1+ Mode O1 1| Atom | δx | δy | δz |
| O1 | 0.035694 | 0.035694 | 0.000000 |
| O1_2 | 0.000000 | 0.000000 | 0.035694 |
| O1_3 | -0.035694 | 0.035694 | 0.000000 |
Irrep GM3+
GM3+ Mode O1 1| Atom | δx | δy | δz |
| O1 | 0.025239 | 0.025239 | 0.000000 |
| O1_2 | 0.000000 | 0.000000 | -0.050479 |
| O1_3 | -0.025239 | 0.025239 | 0.000000 |
Irrep GM4+
GM4+ Mode O1 1| Atom | δx | δy | δz |
| O1 | 0.000000 | 0.000000 | -0.030912 |
| O1_2 | 0.061824 | 0.000000 | 0.000000 |
| O1_3 | 0.000000 | 0.000000 | 0.030912 |
Irrep GM5+
GM5+ Mode Sr1 1| Atom | δx | δy | δz |
| Sr1 | 0.087432 | 0.000000 | 0.000000 |
GM5+ Mode Sr1 2
| Atom | δx | δy | δz |
| Sr1 | 0.000000 | 0.000000 | -0.061824 |
GM5+ Mode O1 1
| Atom | δx | δy | δz |
| O1 | 0.043716 | -0.043716 | 0.000000 |
| O1_2 | 0.000000 | 0.000000 | 0.000000 |
| O1_3 | -0.043716 | -0.043716 | 0.000000 |
GM5+ Mode O1 2
| Atom | δx | δy | δz |
| O1 | 0.000000 | 0.000000 | -0.030912 |
| O1_2 | -0.061824 | 0.000000 | 0.000000 |
| O1_3 | 0.000000 | 0.000000 | 0.030912 |
K-vector: X = (0,1,0)
Irrep X2+
X2+ Mode O1 1| Atom | δx | δy | δz |
| O1 | -0.043716 | -0.043716 | 0.000000 |
| O1_2 | 0.000000 | 0.000000 | 0.000000 |
| O1_3 | -0.043716 | 0.043716 | 0.000000 |
Irrep X3+
X3+ Mode O1 1| Atom | δx | δy | δz |
| O1 | -0.043716 | 0.043716 | 0.000000 |
| O1_2 | 0.000000 | 0.000000 | 0.000000 |
| O1_3 | -0.043716 | -0.043716 | 0.000000 |
Irrep X5+
X5+ Mode Sr1 1| Atom | δx | δy | δz |
| Sr1 | 0.000000 | 0.087432 | 0.000000 |
X5+ Mode O1 1
| Atom | δx | δy | δz |
| O1 | 0.000000 | 0.000000 | 0.000000 |
| O1_2 | 0.000000 | -0.087432 | 0.000000 |
| O1_3 | 0.000000 | 0.000000 | 0.000000 |
X5+ Mode O1 2
| Atom | δx | δy | δz |
| O1 | 0.000000 | 0.000000 | 0.043716 |
| O1_2 | 0.000000 | 0.000000 | 0.000000 |
| O1_3 | 0.000000 | 0.000000 | 0.043716 |
Hide Normalized Symmetry Modes Show Normalized Symmetry Modes
K-vector: GM = (0,0,0)
Irrep: GM1+
Direction: (a)
Isotropy Subgroup: 225 Fm-3m Oh-5
Transformation matrix:[ 1 0 0 ] [ 0] [ 0 1 0 ] [ 0] [ 0 0 1 ] [ 0]
The amplitude of this distortion is:
AGM1+= 0.2423 Å
There is one mode with this symmetry, and its amplitude in Ångströms is:
| O1 1 |
| -0.2423 |
Normalized polarization vector expressed as displacements (in cell relative units) of the atoms in the asymmetric unit of the structure:(normalization unit: 1 Ångström)
| Atom | δx | δy | δz |
| Sb1 | 0.0000 | 0.0000 | 0.0000 |
| Sc1 | 0.0000 | 0.0000 | 0.0000 |
| Sr1 | 0.0000 | 0.0000 | 0.0000 |
| O1 | -0.0357 | -0.0357 | 0.0000 |
| O1_2 | 0.0000 | 0.0000 | -0.0357 |
| O1_3 | 0.0357 | -0.0357 | 0.0000 |
Irrep: GM3+
Direction: (a,0)
Isotropy Subgroup: 139 I4/mmm D4h-17
Transformation matrix:[ 1/2 -1/2 0 ] [ 0] [ 1/2 1/2 0 ] [ 0] [ 0 0 1 ] [ 0]
The amplitude of this distortion is:
AGM3+= 0.0030 Å
There is one mode with this symmetry, and its amplitude in Ångströms is:
| O1 1 |
| -0.0030 |
Normalized polarization vector expressed as displacements (in cell relative units) of the atoms in the asymmetric unit of the structure:(normalization unit: 1 Ångström)
| Atom | δx | δy | δz |
| Sb1 | 0.0000 | 0.0000 | 0.0000 |
| Sc1 | 0.0000 | 0.0000 | 0.0000 |
| Sr1 | 0.0000 | 0.0000 | 0.0000 |
| O1 | -0.0252 | -0.0252 | 0.0000 |
| O1_2 | 0.0000 | 0.0000 | 0.0505 |
| O1_3 | 0.0252 | -0.0252 | 0.0000 |
Irrep: GM4+
Direction: (a,a,0)
Isotropy Subgroup: 12 C2/m C2h-3
Transformation matrix:[ -1/2 -1/2 1/2 ] [ 0] [ 1/2 -1/2 -1/2 ] [ 0] [ 1 0 0 ] [ 0]
The amplitude of this distortion is:
AGM4+= 0.8225 Å
There is one mode with this symmetry, and its amplitude in Ångströms is:
| O1 1 |
| -0.8225 |
Normalized polarization vector expressed as displacements (in cell relative units) of the atoms in the asymmetric unit of the structure:(normalization unit: 1 Ångström)
| Atom | δx | δy | δz |
| Sb1 | 0.0000 | 0.0000 | 0.0000 |
| Sc1 | 0.0000 | 0.0000 | 0.0000 |
| Sr1 | 0.0000 | 0.0000 | 0.0000 |
| O1 | 0.0000 | 0.0000 | 0.0309 |
| O1_2 | -0.0618 | 0.0000 | 0.0000 |
| O1_3 | 0.0000 | 0.0000 | -0.0309 |
Irrep: GM5+
Direction: (-b,a,-a)
Isotropy Subgroup: 12 C2/m C2h-3
Transformation matrix:[ -1/2 1/2 1/2 ] [ 0] [ 1/2 1/2 -1/2 ] [ 0] [ -1 0 0 ] [ 0]
The amplitude of this distortion is:
AGM5+= 0.0491 Å
Normalized polarization vector: (in terms of the amplitudes of the (normalized) atomic symmetry modes)
| Sr1 1 | Sr1 2 | O1 1 | O1 2 |
| 0.4896 | 0.4286 | -0.7577 | -0.0495 |
| NOTE: | A second number next to the label counts the different symmetry modes that may happen for that orbit. |
Normalized polarization vector expressed as displacements (in cell relative units) of the atoms in the asymmetric unit of the structure:(normalization unit: 1 Ångström)
| Atom | δx | δy | δz |
| Sb1 | 0.0000 | 0.0000 | 0.0000 |
| Sc1 | 0.0000 | 0.0000 | 0.0000 |
| Sr1 | 0.0428 | 0.0000 | -0.0265 |
| O1 | -0.0331 | 0.0331 | 0.0015 |
| O1_2 | 0.0031 | 0.0000 | 0.0000 |
| O1_3 | 0.0331 | 0.0331 | -0.0015 |
K-vector: X = (0,1,0)
Irrep: X2+
Direction: (0,a,0)
Isotropy Subgroup: 136 P4_2/mnm D4h-14
Transformation matrix:[ 1/2 1/2 0 ] [ 0] [ -1/2 1/2 0 ] [ 0] [ 0 0 1 ] [ 0]
The amplitude of this distortion is:
AX2+= 0.0315 Å
There is one mode with this symmetry, and its amplitude in Ångströms is:
| O1 1 |
| 0.0315 |
Normalized polarization vector expressed as displacements (in cell relative units) of the atoms in the asymmetric unit of the structure:(normalization unit: 1 Ångström)
| Atom | δx | δy | δz |
| Sb1 | 0.0000 | 0.0000 | 0.0000 |
| Sc1 | 0.0000 | 0.0000 | 0.0000 |
| Sr1 | 0.0000 | 0.0000 | 0.0000 |
| O1 | -0.0437 | -0.0437 | 0.0000 |
| O1_2 | 0.0000 | 0.0000 | 0.0000 |
| O1_3 | -0.0437 | 0.0437 | 0.0000 |
Irrep: X3+
Direction: (0,a,0)
Isotropy Subgroup: 128 P4/mnc D4h-6
Transformation matrix:[ 1/2 1/2 0 ] [ 0] [ -1/2 1/2 0 ] [ 0] [ 0 0 1 ] [ 0]
The amplitude of this distortion is:
AX3+= 0.3746 Å
There is one mode with this symmetry, and its amplitude in Ångströms is:
| O1 1 |
| 0.3746 |
Normalized polarization vector expressed as displacements (in cell relative units) of the atoms in the asymmetric unit of the structure:(normalization unit: 1 Ångström)
| Atom | δx | δy | δz |
| Sb1 | 0.0000 | 0.0000 | 0.0000 |
| Sc1 | 0.0000 | 0.0000 | 0.0000 |
| Sr1 | 0.0000 | 0.0000 | 0.0000 |
| O1 | -0.0437 | 0.0437 | 0.0000 |
| O1_2 | 0.0000 | 0.0000 | 0.0000 |
| O1_3 | -0.0437 | -0.0437 | 0.0000 |
Irrep: X5+
Direction: (a,a,0,0,a,-a)
Isotropy Subgroup: 58 Pnnm D2h-12
Transformation matrix:[ 0 1/2 -1/2 ] [ 0] [ 0 1/2 1/2 ] [ 0] [ 1 0 0 ] [ 0]
The amplitude of this distortion is:
AX5+= 0.1421 Å
Normalized polarization vector: (in terms of the amplitudes of the (normalized) atomic symmetry modes)
| Sr1 1 | O1 1 | O1 2 |
| 0.7968 | -0.4507 | -0.4024 |
| NOTE: | A second number next to the label counts the different symmetry modes that may happen for that orbit. |
Normalized polarization vector expressed as displacements (in cell relative units) of the atoms in the asymmetric unit of the structure:(normalization unit: 1 Ångström)
| Atom | δx | δy | δz |
| Sb1 | 0.0000 | 0.0000 | 0.0000 |
| Sc1 | 0.0000 | 0.0000 | 0.0000 |
| Sr1 | 0.0000 | 0.0697 | 0.0000 |
| O1 | 0.0000 | 0.0000 | -0.0176 |
| O1_2 | 0.0000 | 0.0394 | 0.0000 |
| O1_3 | 0.0000 | 0.0000 | -0.0176 |
Hide Detailed information
|
For comments, please mail to administrador.bcs@ehu.eus |
